
Home > Research Teams > Structure and Dynamics of Isolated Complex and Photoexcited Systems > Molécules Agrégats Neutres Ionisés ou Protonés (MANIP) > Spectroscopie et dynamique des états excités de systèmes moléculaires protonés
Spectroscopie et dynamique des états excités de systèmes moléculaires protonés
Spectroscopie et dynamique multi-échelle à 10 K
by - 16 February 2018 (modifié le 17 February 2018)
Le dispositif Ions Froids permet de former des ions et de les piéger à une température d’une dizaine de kelvin. Les espèces piégées se trouvent alors dans le niveau vibrationnel fondamental à l’état électronique fondamental. Nous pouvons accéder à la spectroscopie UV résolue vibrationnellement et obtenir des informations sur la structure par les techniques usuelles de spectroscopie de double résonances.
L’évolution des rapports de branchement en fonction de l’excès d’énergie à l’état excité peut être suivie grâce à une détection multiplexée par spectrométrie de masse à temps de vol. La mesure des durées de vie résolue en conformation et en énergie renseigne sur les voies de désactivation à l’état excité.
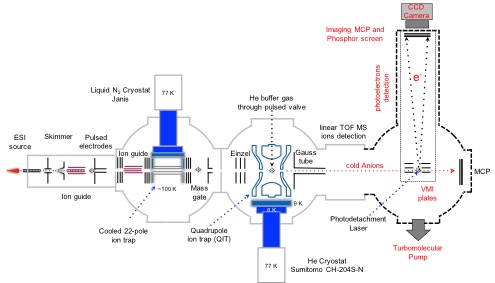
Dynamique multi-échelle
La spécificité du dispositif expérimental Ions Froids combiné aux différentes sources laser du Serveur Laser au CLUPS repose sur l’application de la technique pompe-sonde sur des échelles de temps allant de la subpicoseconde (< 10−12 s) à la dizaine de millisecondes (> 10−3 s). Nous avons récemment démontré qu’il est possible de suivre les étapes de la dynamique de relaxation des états excités jusqu’au retour à l’état fondamental. Nous sommes les seuls au monde à faire ce type de mesures résolues en temps et en énergie sur des espèces refroidies en trappe à ions cryogénique.
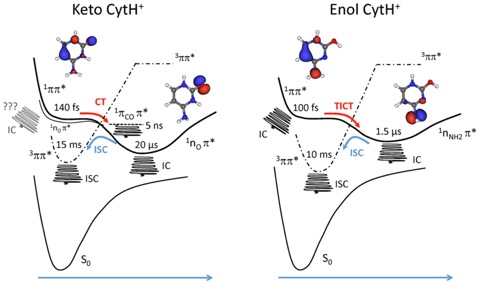
Dans le cas des bases de l’ADN, les intersections coniques peuvent fournir des voies de relaxation rapides. Les mesures pompe-sonde à l’échelle de la femtoseconde sur les bases de l’ADN ont permis de caractériser l’étape primaire de la dynamique de relaxation de l’état localement excité qui suit une excitation électronique. Pour la cytosine protonée, nous avons récemment démontré que la dynamique de relaxation peut être totalement suivie sur une échelle allant de la subpicoseconde à la dizaine de millisecondes. Dans ce système, la durée de vie de l’état localement excité est très courte (d’une centaine de femtosecondes) et le déclin s’opère via des états à transfert de charge et triplet à longue durée de vie, microsecondes et millisecondes, respectivement. Un mécanisme en trois étapes (1ππ∗ → 1CT → 3ππ∗) est proposé, où la conversion interne de chaque état peut se produire menant finalement à la fragmentation à l’état fondamental (figure ci-dessus).
Cette étude sur la cytosine protonée isolée nous montre qu’une frac- tion non-négligeable de la population dans l’état localement excité 1ππ∗ ne retourne pas à l’état fondamental mais va explorer d’autres états, en particulier l’état triplet. Dans ces conditions où les espèces sont dépourvues de tout effet macroscopique dû au solvant, le processus de relaxation s’opère sur des échelles de temps bien au delà de la dizaine de millisecondes. Nous avons une vision la plus complète possible de la dynamique de relaxation.
Spectroscopie de type UV-UV hole-burning
La Tyramine protonée est le système modèle où le groupement -COOH sur le Cα dans la Tyrosine est remplacé par un atome d’hydrogène. Le groupement NH3+ se trouve au dessus du cycle aromatique (analogue des conformères rot de la tyrosine protonée) dans les deux conformations de plus basse énergie, qui ne diffèrent alors que par l’orientation du groupement hydroxyle (syn ou anti) par rapport au groupement NH3+.
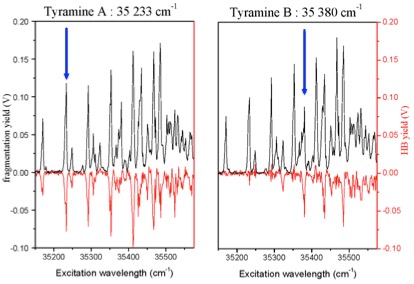
La présence de ces deux conformères à basse température dans le piège cryogénique a été mis en évidence par des expériences de type UV-UV hole-burning, combinées aux calculs ab initio et une analyse Franck-Condon des spectres vibroniques (figure ci-dessus). Avec cette méthode originale de dépopulation par un laser UV, la discrimination des conformères syn et anti peut être réalisée, ce qui n’est pas faisable dans le cas de la Tyrosine protonée en employant un laser IR de dépopulation car l’écart énergétique est trop faible (la différence d’énergie est seulement de 0,5 cm−1 pour le mode d’élongation N−H lié).
La voie de fragmentation prépondérante est la perte d’ammoniac (fragment m/z 121), tout comme dans les expériences de dissociations induites par collisions. La rupture de la liaison Cα−Cβ (fragments m/z 108 et 109) apparaît, quant à elle, dès la transition 0-0 mais celle-ci est au moins dix fois moins intense que la perte d’ammoniac. Ces processus de désactivation contrastent avec la Tyrosine protonée, où la rupture Cα−Cβ est la voie de fragmentation prépondérante autour de l’origine de la transition ππ∗. L’ion m/z 108 est formé après transfert à l’état excité d’un proton du groupement NH3+ vers le cycle aromatique comme dans le cas des molécules protonées : la phényléthylamine, la tyrosine et la phénylalanine. Le fragment m/z 109 est formé à la suite d’un transfert de proton et d’un transfert d’hydrogène comme il a déjà été observé dans le cas de molécules protonées (tyramine, tryptophane et tryptamine) après excitation à 263 nm.
Collaborations :
![]() Ruxandra Gref (ISMO, Université Paris-Sud)
Ruxandra Gref (ISMO, Université Paris-Sud)
![]() Fabien Gatti (ISMO, Université Paris-Sud)
Fabien Gatti (ISMO, Université Paris-Sud)
![]() Christophe Jouvet et Claude Dedonder-Lardeux (PIIM, Aix-Marseille Université)
Christophe Jouvet et Claude Dedonder-Lardeux (PIIM, Aix-Marseille Université)
![]() Mario Barbatti (ICR, Aix-Marseille Université)
Mario Barbatti (ICR, Aix-Marseille Université)
![]() Nicolas Nieuwjaer, Frédéric Lecomte, Bruno Manil et Charles Desfrançois (LPL, Université Paris 13)
Nicolas Nieuwjaer, Frédéric Lecomte, Bruno Manil et Charles Desfrançois (LPL, Université Paris 13)
![]() Luke MacAleese (ILM, Université Claude Bernard Lyon 1)
Luke MacAleese (ILM, Université Claude Bernard Lyon 1)
![]() Gustavo Pino (Universidad Nacional de Cordoba, Argentine)
Gustavo Pino (Universidad Nacional de Cordoba, Argentine)
![]() Enersto Marceca (Universidad de Buenos Aires, Argentine)
Enersto Marceca (Universidad de Buenos Aires, Argentine)
![]() Masaaki Fujii (Tokyo Institut of Technology, Japon)
Masaaki Fujii (Tokyo Institut of Technology, Japon)